

Inhibiting SARS-CoV-2 Main Protease
The development of a new drug is a costly and lengthy process. Today as the world is facing a pandemic there is an urgent need to identify quickly drugs that stop virus proliferation. Drug repurposing offers an attractive alternative to this lengthy process by trying to identify if a drug known to be safe in humans could be used to treat new diseases.
While using such repositioned drugs individually might eventually not result in a significant clinical benefit, careful combination of drugs targeting several proteins crucial for virus replication and proliferation could be very effective, as was the case with HIV in the 1990s. The urgent question is which combination would be more effective?
Here we try to understand the structure of the SARS-CoV-2 protease active site, comparing it with existing structures of SARS-CoV protease complexed with micro-molar inhibitors, so that we can better understand the key interactions required to create a good inhibitor for SARS-CoV-2 protease.
We then conducted a virtual screening experiment using a library of FDA-approved drugs to see if some of these are predicted to bind to the protease. We examined how they are predicted to bind to the SARS-CoV-2 protease and could thus be used in a combination therapy.
SARS-CoV–2 Proteins
The SARS-CoV–2 genome from ill patients was quickly isolated and sequenced, providing the sequences of possible protein targets. These proteins share high sequence similarity with SARS-CoV proteins and initially research groups started building homology models. Now we are seeing more and more of these experimentally derived (X-ray and Cryo-EM) structures become available.
One of the best-characterized drug targets among coronaviruses is the main protease: Mpro, also called 3CL Protease.1 Along with the papain-like proteases, this enzyme is essential for processing the polyproteins that are translated from the viral RNA.2 It cleaves the amino acid backbone at 11 cleavage sites on the large polyprotein 1ab (Fig. 1).
There many structures of SARS-CoV Mpro complexed with ligands available at the PDB and the ligands that have a micro molar binding affinity are all covalent ligands.
The active sites of the main protease are highly conserved amongst coronaviruses and are usually composed of four sites (S1′, S1, S2 and S4)3 (Fig. 4).
In the case of PDB structures 2ZU4 and 2GX4 for which ligands have respectively Ki values of 0.038 μM and 0.053 μM, the thiol of cysteine 145 in the S1′ site contacts inhibitors with a covalent bond. When compared with other inhibitors with lower affinity, this seems to be important for higher affinity.
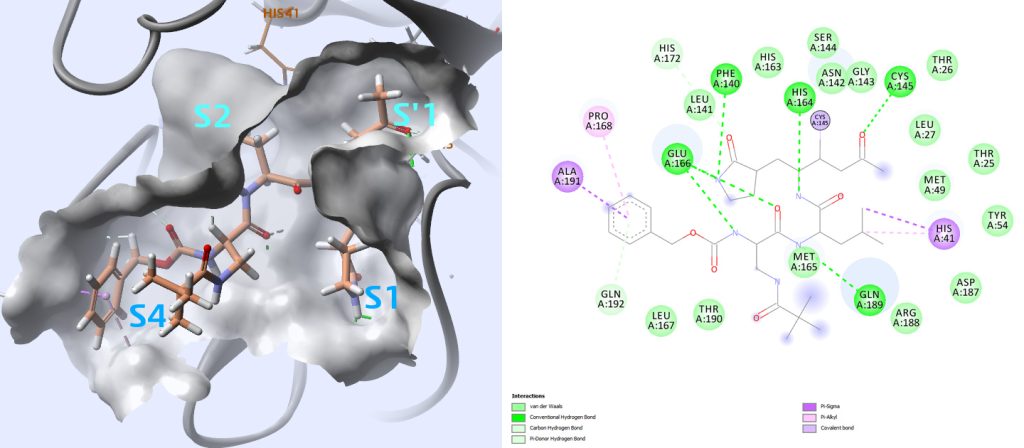
Careful examination of how all these ligands interact with this protease may provide information about key interactions to monitor when analyzing docking results.
Virtual Screening
Taking advantage of a high-resolution crystal structure of SARS-COV-2 protease dimer in complex with a covalently bonded peptide-like inhibitor N3 released in February (6LU7)4, we used this structure to conduct a virtual screening experiment. The inhibitor N3 that is co-crystalized binds in the substrate-binding pocket in an extended conformation. (Fig. 5)
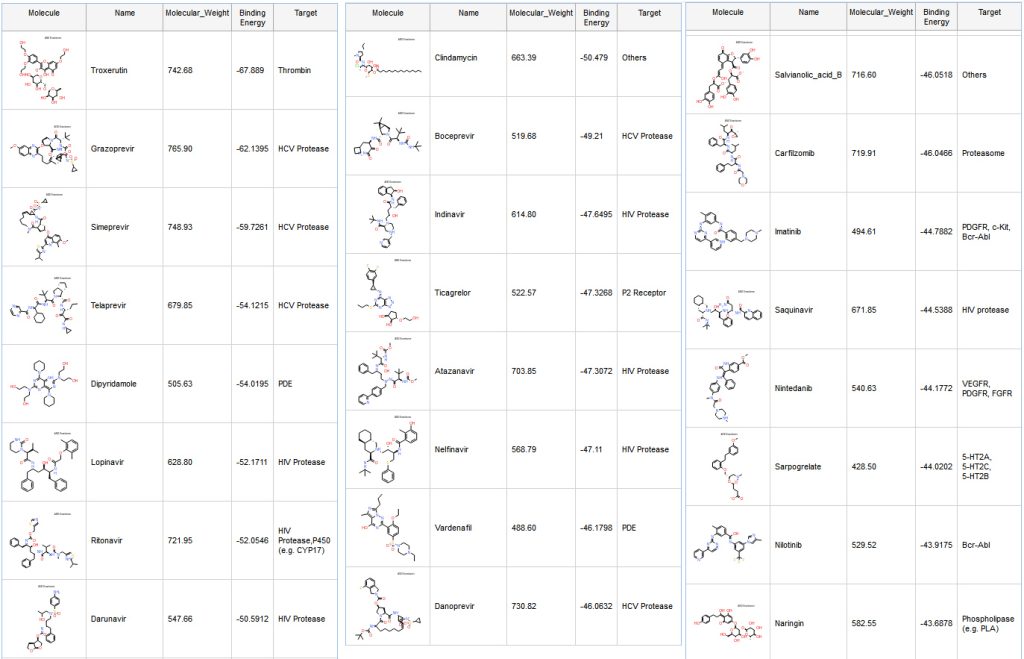
Dipyridamole (Fig. 7), which is the fifth ranked compound, has been mentioned recently in a preprint Therapeutic effects of dipyridamole on COVID-19 patients with coagulation dysfunction, and shown to suppress HCoV-19 replication at an EC50 of 100 nM in vitro.

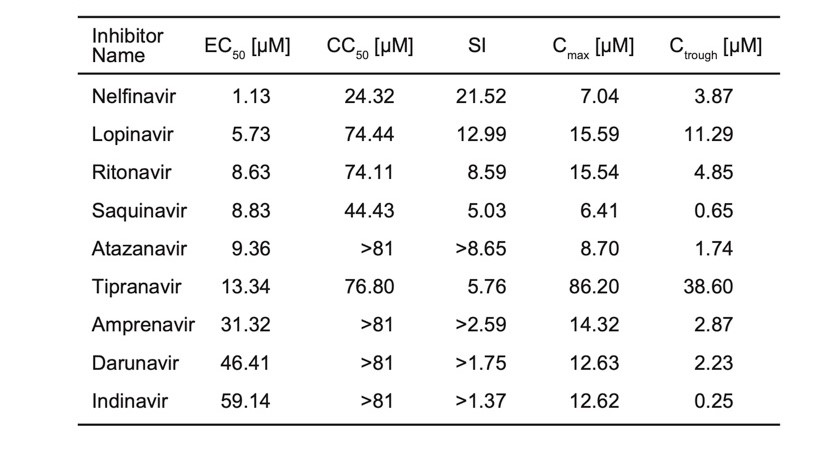
We are reporting the table from the preprint (Table 1). These are not IC50, but EC50, so it does not tell how well these molecules bind to the protease. However, this shows that all those compounds are inhibiting replication of SARS-CoV-2 and some are more active than others.
Ritonavir and Lopinavir are interacting with many residues previously seen in other compounds targeting SARS-CoV protease (Fig. 8 and 9). Although several clinical trials are ongoing with a combination of those two molecules, there are unfortunately no IC50 data available for Ritonavir or Lopinavir on SARS-CoV-2.
A second preprint shows that Atazanavir is active on SARS-CoV-2 infected cells: “Atazanavir inhibits SARS-CoV-2 replication and pro-inflammatory cytokine production.”
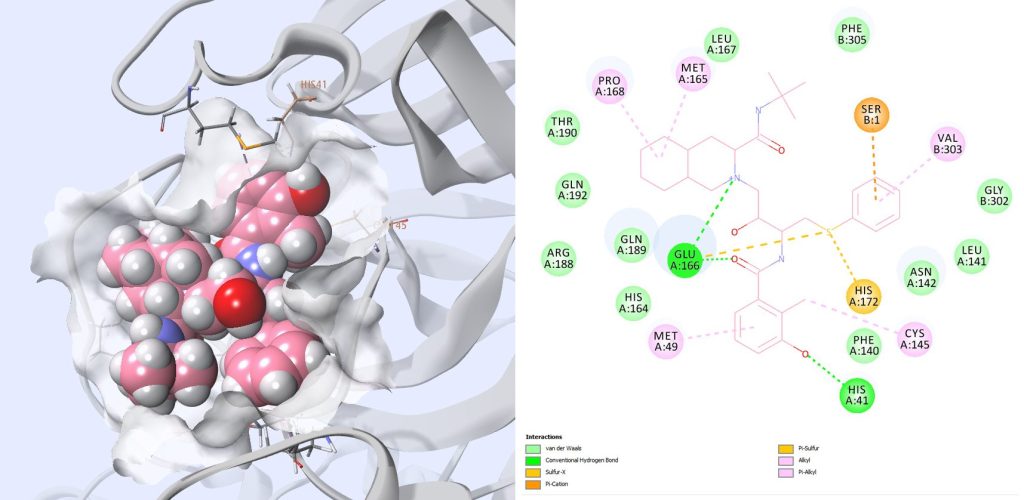
A PDB structure 6W63 that contains a new inhibitor for SARS-CoV-2 was published after we started this work (Fig. 11). There is no published information about the binding affinity of this compound to the SARS-CoV-2 Mpro. The side chains in the binding site are almost in the same orientation as for 6LU7. We docked our compounds and rescored them with MMGBSA approaches as previously and found very similar results.
) for permission to use GOLD for the virtual screening in this work. GOLD has proven success in virtual screening, lead optimization, and identifying the correct binding mode of active molecules. An interface to GOLD is available within Discovery Studio.
References
- K. Anand, J. Ziebuhr, P. Wadhwani, J. R. Mesters, R. Hilgenfeld. Coronavirus main proteinase (3CLpro) structure: Basis for design of anti-SARS drugs. Science. 2003, 300(5626):1763-7.
- R. Hilgenfeld. From SARS to MERS: Crystallographic studies on coronaviral proteases enable antiviral drug design. FEBS J. 2014, 281(18):4085-96.
- H. Yang et al. Design of wide-spectrum inhibitors targeting coronavirus main proteases. PLoS Biol. 2005, 3(10):e324.
- Jin, Z. et al. Structure of Mpro from COVID-19 virus and discovery of its inhibitors. Nature. 2020, doi: 10.1038/s41586-020-2223-y.
- Jo, S. et al. Inhibition of SARS-CoV 3CL protease by flavonoids J Enzyme Inhib Med Chem 2020 Dec;35(1):145-151. doi: 10.1080/14756366.2019.1690480