
By now, we can safely classify the field of NMR crystallography as mature. In fact, it can already celebrate an anniversary – it has been ten years since the International Union of Crystallography recognized its importance by establishing a dedicated commission on NMR Crystallography and Related Methods. The IUCr considers the combined use of experimental and computational techniques as the modern way of understanding the structure and dynamics of solids on an atomic scale.
Growing Power of NMR Crystallography
NMR spectroscopy is uniquely sensitive to the details of short-range structure of materials. The measured values of chemical shift, electric field gradient, J-coupling, help to quantify structural disorder and local distortions caused by defects. This information is a great supplement to the traditional diffraction-based crystallography that looks at the long-range order in crystals. Computing and atomistic modeling is an essential element of the success of NMR crystallography. Experimental spectra are of course valuable, but the true power of the method comes from signal assignments generated in quantum-mechanical calculations. In the world of experimentation, there are more and more powerful permanent magnets in laboratories around the world. To mention just a few, there is a1.2 GHz and 28 Tesla spectrometer at the ETH Zurich, 1 GHz and 23.5 T at University of Warwick (with a new 1.2 GHz machine in procurement), ultra-high field solid state NMR (1.5 GHz, 36 T) at the National High Magnetic Field Laboratory, NHMFL, in Tallahassee, Fl, 1 GHz and 22 T in Grenoble . Such established manufacturers as Bruker and Agilent constantly improve experimental designs. The most powerful machines are extremely expensive – for example, the Warwick installation is based on a £17m grant from the UK government.

Advantages of NMR Crystallography
So the experiments are complex and expensive; this highlights the need to extract every last bit of useful information from the results. Here computations come to the rescue. Solid-state NMR calculations were introduced as far back as 2001 when Pickard and Mauri solved the gauge-invariance problem in periodic boundary conditions [1]; this paper now has nearly 2000 citations. UK government provides dedicated funding to a Collaborative Computational Project for NMR Crystallography, the place that collects software and data relevant to this field. BIOVIA Materials Studio contains a CASTEP NMR module which is currently used to generate most of the first-principles results for solid-state NMR, although other electronic structure packages are getting increasingly interested in implemented the functionality.
OK, so what are the recent exciting applications of NMR crystallography? Let us look at two different examples, one dealing with inorganic materials for solid oxide fuel cells (SOFCs), and another dedicated to the characterization of molecular crystals in pharmaceutical applications.
SOFC electrolytes must satisfy several requirements including fast ion conductivity at moderate temperatures (e.g., less than 800 °C to 1000 °C range of yttria-stabilized zirconia). An international team from the University of Liverpool and from NHMFL investigated the structure and properties of materials from the melilite family, LaSrGa3O7 [2]. This layered material is normally distorted because of local disorder in cationic and anionic layers. The structure of the La-doped phase is promising for good oxygen transport in the temperature range 600 °C to 800 °C. It seems that the essential structural feature responsible for the high mobility of oxygen is the presence of three distinct Ga-O structural units. La-doped material has four-connected Ga(1)O4, three-connected Ga(2)O4 and Ga(2’)O5. The experiment required the highest possible magnetic field of 35.2 T to resolve the signals from different Ga ions. And all one can really see in the recorded 71Ga spectra is the appearance of two additional and rather weak peaks in the La-doped sample compared to the stoichiometric one. A deeper understanding of the nature of those changes comes from CASTEP NMR results that explain the origin of each additional peak.
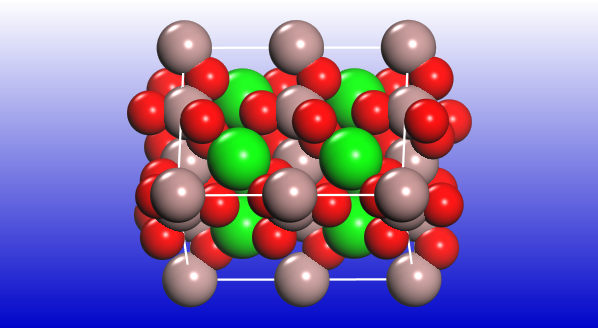
Overall, this is an impressive case that combines the most powerful experimental setup available to date anywhere in the world with the robust computational tool to decode structural details hidden in NMR spectra.
Inorganic materials such as solid-state electrolytes for fuel elements or novel batteries often exhibit complex chemistry and combine multiple elements in unusual combinations (e.g., Ga and La quadrupole nuclei in the SOFC example above). Pharmaceutical applications present a different set of complications to NMR crystallographers. Here the elements are well defined in advance (C, O, H, and N would normally cover nearly all interesting combinations). The crystal packing and polymorphism, on the other hand, complicate things. Once again, the power of the method is in combining experiment and calculation. NMR chemical shifts provide such details as the number of molecules in the asymmetric unit, the information of great value for crystal structure prediction workflows. 1H, 13C, and 15N chemical shifts are sensitive to noncovalent interactions such as hydrogen bonds, they can be used to characterize disorder, to distinguish between salts and cocrystals, etc.
Scientists at the Warwick University NMR centre jointly with the researchers of AstraZeneca worked for many years on improved usability of the available tools. They produced a toolbox that is used extensively in the CCP-NC workflows [3]. The authors used BIOVIA Materials Studio powerful scripting layer to automate the calculations, extract the data and visualize the results. Such developments paved the way to recent large-sale studies that push the boundaries of NMR crystallography applications even further.
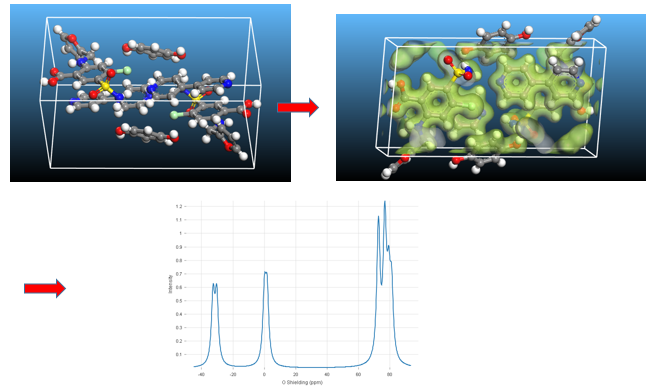
A team from AstraZeneca and EPFL tackled a seemingly impossible task: they applied the principles that work so well for ordered crystalline phases to solve the structure of amorphous molecular solids. Their paper combines traditional atomistic modeling with a machine learning approach [4]. The conventional method computes chemical shifts for specific structures and compares the results to the experiment. This is not going to work for large amorphous systems for the simple reason of prohibitive computational cost. The team from EPFL developed a machine learning model ShiftML2 that is trained on DFT results for thousands of systems with different chemical environments and describes both ideal and distorted structures. This model is combined with solid-state NMR experiments and with classical molecular dynamics simulations [4]. By considering the combined shifts of all 1H and 13C atomic sites in the molecule, the team determined the structure of the amorphous form of the drug AZD4625. Remarkably, prior to this study the crystal structure even of the pure compound was not known.
The combined team from AstraZeneca, C4X Discovery, and Warwick University [5] illustrates a different combination of NMR crystallography and data science tools. Here the problem to solve was – how to discriminate between crystalline polymorphs of flexible molecules. Crystals such as furosemide are formed by the packing of molecules with multiple conformational degrees of freedom. As the authors state in the paper, unwanted polymorphs of active pharmaceutical ingredients, APIs, can be close in energy to the desired form and they continuously emerge in the fabrication process. The risks can be somewhat reduced if there is a clear way of distinguishing and characterizing such polymorphs. The path towards such derisking can be based on the regression analysis of experimental and calculated NMR data for solution and solid-state forms of the molecules of interest. The paper used Monte Carlo random sampling of the structure of the solution to calculate the solution-state chemical shift, and to compare measured and calculated differences between solution and solid form.
There is no doubt that the last two approaches ([4] and [5]) will be unified in the very near future. ML-based chemical shifts can be used instead of the DFT-calculated data for special configurations of the solvated molecule, to reduce overall computational cost by orders of magnitude.
NMR crystallography clearly came of age in the last couple of decades – I would no longer call it “an emerging field” as it is characterized on the CCP-NC website. Even this brief summary of recent applications shows the breadth and relevance of solid-state NMR to design and characterization of materials for novel batteries, fuel cells, and pharmaceutics. There is a feedback loop where an appetite for novel applications drives the manufacture of new and more powerful experimental machines, the establishment of numerous national NMR centers, and the development of new computational approaches. DFT calculations with tools like BIOVIA CASTEP remain the cornerstone of theoretical studies. The DFT data can now be collected in databases – CCP-NC provides one example – and used for training machine-learning models. The field rapidly expands and represents an embodiment of the Dassault Systèmes V+R philosophy in action: the symbiosis between virtual (simulations and machine learning) and real (NMR experiments). Computational experiments are not meant to replace physical ones, especially when the two can work so well together!
Finally, here are two announcements of the upcoming events of interest to NMR crystallography community.
- The 2024 SMARTER meeting on NMR Crystallography (Sept 2024, Aveiro, Portugal) brings together scientists in the areas of computation, diffraction, magnetic resonance and other spectroscopic techniques to promote and develop SMARTER approaches to optimally combine various techniques for structure elucidation of complex inorganic, organic and biological materials. The website of the event is available here . The following invited speakers have already confirmed their presence at the University of Aveiro: Bradley Chmelka (UC Santa Barbara), Michal Leskes (Weizmann Institute), Ute Kolb (Johannes Gutenberg University Mainz) and Zhehao Huang (Stockholm University).
- Oxford will host an advanced masterclass in CASTEP NMR calculations from 8th-10th July 2024. The event has limited places and will prioritize UK-based applicants. Organizers expect participants to have a reasonable level of experience with NMR calculations with CASTEP, and to be using it as part of their current research. The application form is here: Topics to be covered include:
- GIPAW theory of NMR parameters in solids (shielding, EFG, J)Relativistic effects on NMR parameters (heavy atoms)Running efficiently on parallel machines
- Visualisation and post-processing tools (e.g. MagresView, MagresPython, NICS)
References
- Pickard & Mauri, Phys. Rev. B 63, 245101 (2001) https://doi.org/10.1103/PhysRevB.63.245101
- Corto et al., ChemPhysChem, 25,e202300934 (2024) https://doi.org/10.1002/cphc.202300934
- Szell et al., Solid State Nucl. Mag. Res. 116, 101761 (2021) https://doi.org/10.1016/j.ssnmr.2021.101761
- Cordova et al., Nature Communications, 14, 5138 (2023) https://doi.org/10.1038/s41467-023-40853-2)
- Rahman et al., J. Phys. Chem. A, 128, 1793, 2024 https://doi.org/10.1021/acs.jpca.3c07732

Dr. Victor Milman is a BIOVIA R&D Scientific Application Director and Head of BIOVIA Science Council. With a doctorate in solid state physics from The Ukrainian Academy of Sciences, Dr. Milman has contributed to the development of CASTEP, a revolutionary code for quantum-mechanical modeling of materials and has over 200 peer-reviewed publications with an h-index of 44.