In the ever-evolving landscape of drug discovery, the chemistry of medicines is racing ahead of the words we use to describe it. Antisense oligonucleotides, mRNA vaccines, therapeutic peptides, antibody-drug conjugates, PROTACs — every one of these modalities is built from chemical building blocks that didn’t exist in standard catalogues a decade ago. The pace of what we can make has outpaced the pace at which we can name it. And in a world where data exchange depends on naming, that gap is no longer an aesthetic concern. It is the bottleneck.
The Naming Bottleneck: When Curation Cannot Keep Up
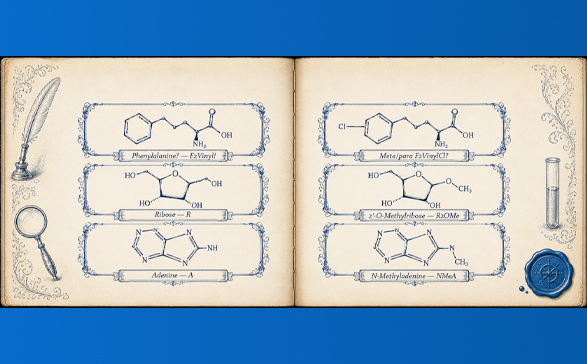
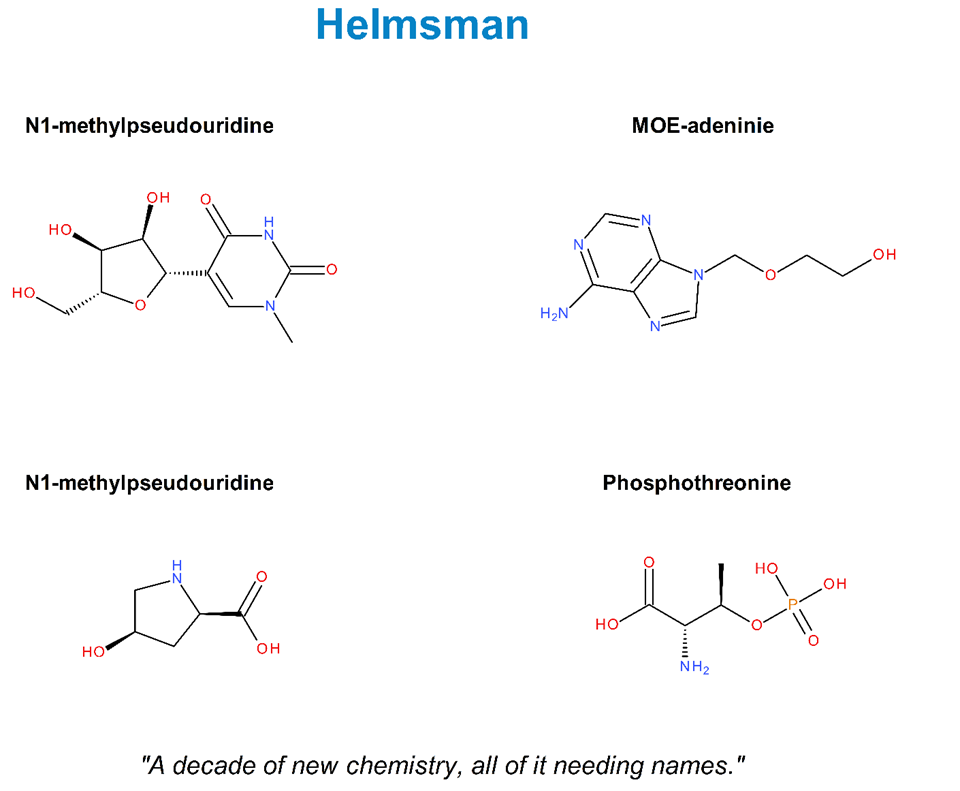
The chemistry that has reshaped therapeutic development in the last decade is, structurally, modified residues at scale. The N1-methylpseudouridine (m1Ψ) in modern mRNA vaccines is a modified nucleobase. The 2′-O-methoxyethyl (MOE) groups in approved antisense oligonucleotides are modified ribose substituents. Therapeutic peptides increasingly carry non-canonical amino acids — α-methyl, β-hydroxy, fused-ring backbones — that fall outside the twenty standard codes. Every one of these residues must enter a registry; every registry entry needs a unique, parseable, audit-trail-bearing name.
The standards exist. Two complementary representations — SCSR (Self-Contained Sequence Representation) and HELM (Hierarchical Editing Language for Macromolecules) — give the field a way to represent any monomer, canonical or not, as a structured composition. What these standards cannot do alone, and were never designed to do, is coin the names for residues no human curator has seen yet. That work has historically been manual: a chemist in a curation team writing a name, another reviewing it, a third propagating it across siblings. It works at the bench. It does not work at library scale.
Agentic AI: A New Mode of Working with Chemistry Software
The starting point for this work is HELMify, a recent and groundbreaking demonstration that a language model can be used to propose HELM-compatible names for monomers the curated catalogue does not cover. Most chemists’ contact with AI so far has been single-turn — a model answering a question, completing a structure, summarizing a document. Agentic AI is something different, and it is worth defining precisely because the term is now everywhere. An agentic workflow is a small team of specialized AI models, each with a defined role, a defined freedom to act, and a defined constraint that bounds it. The models work in concert toward a single, verifiable goal. They iterate. They check each other’s work. And critically, they are gated at every step by a deterministic rule — in chemistry, by structural reality.
The advance over single-turn AI is twofold. First, specialisation: a model asked to perform one well-scoped task — propose a substituent name, audit a proposal, escalate when stuck — outperforms a generalist asked to do everything at once. Second, verifiability: when each model’s output passes through a deterministic structural check before it propagates, the workflow can be bold without being reckless. The model proposes; the structure decides.
BIOVIA Helmsman: An Agentic Approach to Monomer Naming
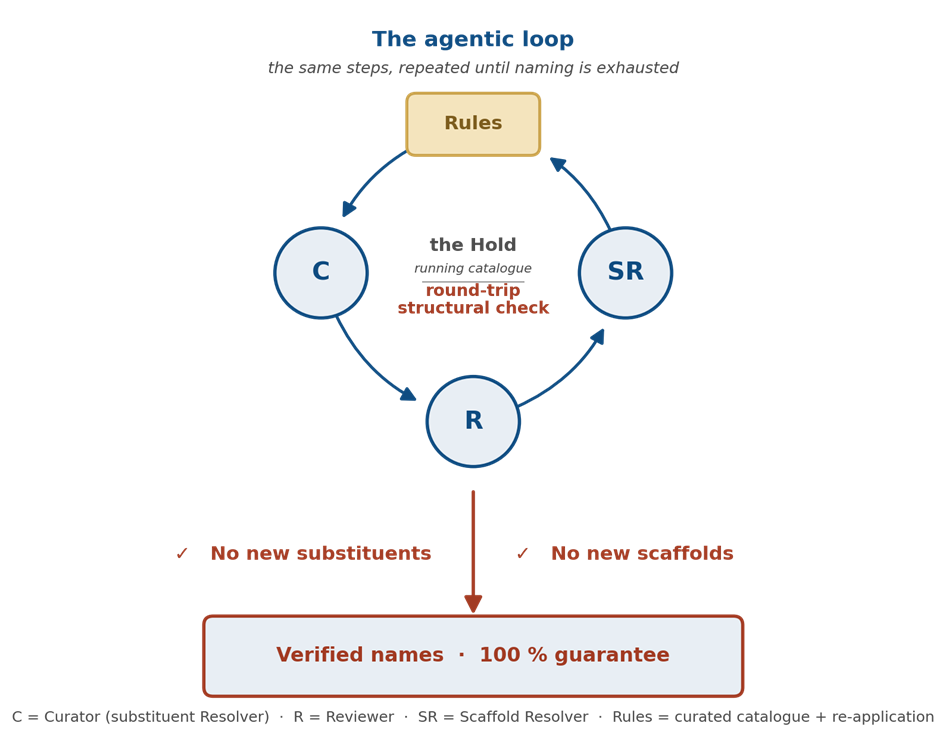
This is the design principle behind BIOVIA Helmsman, an agentic AI pipeline for naming single-monomer residues at library scale. Helmsman does not replace the curated HELM Core Monomer Set or any SCSR template library — a residue that matches a catalogued monomer is returned under its catalogued symbol. Helmsman handles the residues the catalogue does not yet contain. It does so by orchestrating three specialized AI agents and a deterministic rule engine in a single loop, with every proposal verified by reconstructing the molecule the name encodes and comparing it back to the input.
The four participants each have one job:
- The Curator (also called the substituent Resolver) proposes a name for an unnamed substituent group. It works once per distinct piece of chemistry, not once per molecule — a single name proposal, once accepted, propagates across every sibling residue carrying the same group.
- The Reviewer audits each newly named substituent immediately, applying a deliberately skeptical eye. It catches name collisions, near-duplicate vocabulary, and the cardinal error of naming a whole ring system as a “decoration.” It keeps, revises, or removes.
- The Scaffold Resolver handles the residues no amount of substituent naming can fix — the ones where the parent scaffold itself is novel. It re-matches against an extended scaffold library, registers new parent scaffolds when justified, and falls back to whole-molecule identity names when a residue is genuinely irreducible.
- The rules engine propagates every accepted name across the whole library after each step, so a single act of naming benefits every molecule that contains the same chemistry.
These four cycle until two conditions hold simultaneously: there are no new substituents for the Curator to name, and a fresh Scaffold Resolver pass yields no new parent scaffolds. The loop stops only when both gates are clear.
A Worked Example: From Caffeine to a Verified Name
Consider a familiar molecule — caffeine. To a similarity-search fingerprint, caffeine looks remarkably like adenine: same purine skeleton, same fused bicyclic core. A naïve rule engine would dock caffeine against the adenine parent scaffold, only to discover that caffeine carries two ring carbonyls (the 2,6-dione oxidation pattern) that simply cannot be expressed as decorations on a base. The fit fails.
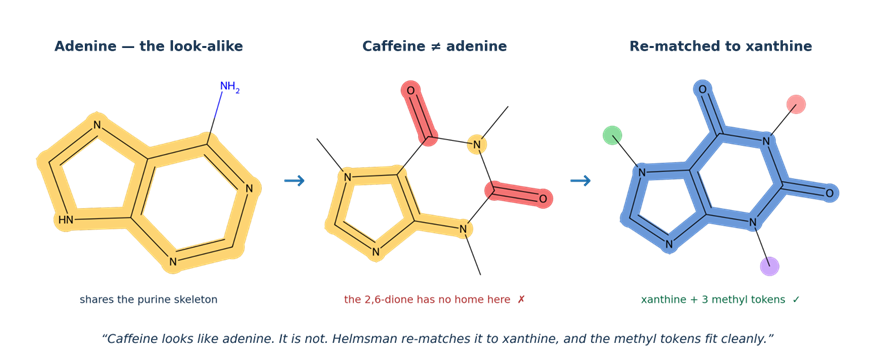
In Helmsman, the Scaffold Resolver asks not “what is this extra carbonyl?” but “did we start from the wrong parent scaffold?” It consults an extended scaffold library, finds xanthine (Xan), and re-matches the molecule. Under xanthine, caffeine names cleanly as Xan2Me10Me6Me — three methyl groups on the xanthine core — with no invented vocabulary required. The structural check confirms: parse the name, reconstruct the molecule, compare to the input. Identical. Across the most recent benchmark, this single re-matching pattern rescued 66 methylxanthine residues from misclassification.
Results: A 100 % Naming Guarantee, Verified by Structure
On a benchmark of 1,352 single-monomer residues drawn from MODOMICS, SwissSidechain, and substructure slices of ChEMBL — all chemistry beyond today’s curated HELM libraries — Helmsman names every input. Every name is verified by structural read-back. The rule engine alone names 478 (35.4 %); the rule engine plus the agentic loop reaches 1,352 of 1,352 (100 %).
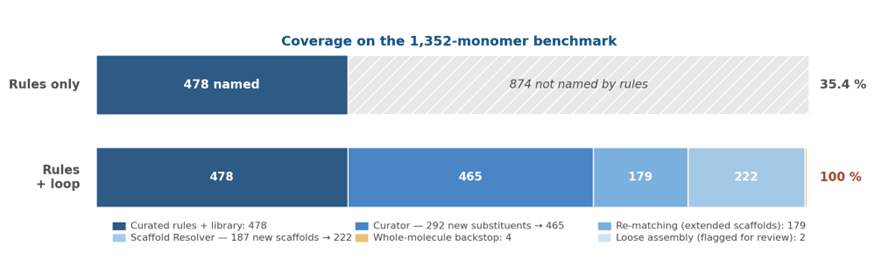
The catalogue itself grows along the way. The Curator adds 292 new substituent names to an exportable vocabulary that nearly triples the original 210-entry curated baseline. The Scaffold Resolver adds 187 new parent scaffolds. Every name carries its origin in a provenance column; every catalogue entry carries the Reviewer’s verdict. The output is registration-grade and auditable end-to-end.
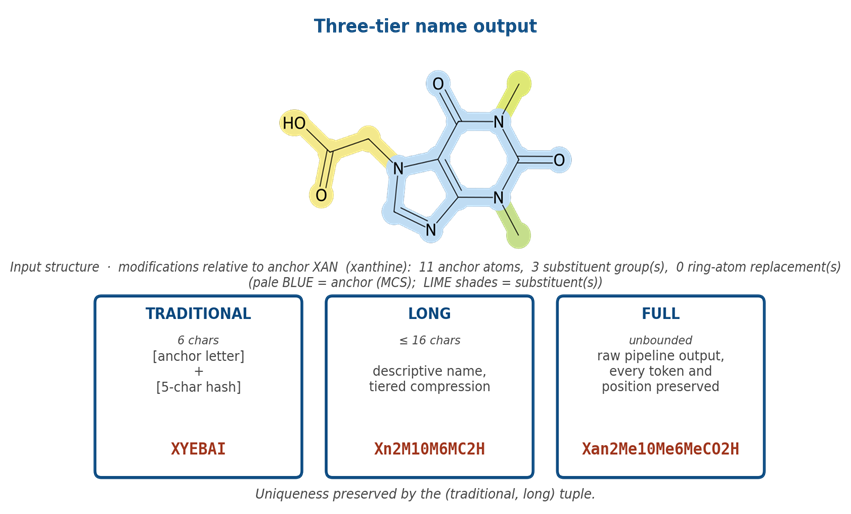
Three name forms per residue: a fixed-width registry identifier, a descriptive long name, and a full canonical name that round-trips through the verifier.
A Catalyst for the Field: Helmsman, Free for Pistoia Alliance Partners
BIOVIA’s commitment to open chemistry standards is long-standing — across SCSR, HELM, and the partnership with the Pistoia Alliance that has anchored two decades of monomer interoperability. Helmsman is the next chapter in that commitment. We are making Helmsman freely available to Pistoia Alliance member organisations as a shared resource for monomer naming, registration, and data exchange.
The decision is deliberate. A naming standard is only as useful as the community that adopts it; a naming pipeline is only as useful as the catalogue it builds. By offering Helmsman openly, BIOVIA is investing in a future in which the chemistry of modern therapeutics — across companies, across libraries, across institutions — speaks a common, verifiable vocabulary. The agentic loop is the engine. The Pistoia Alliance is the place where the field assembles around it.
Navigating What’s Next
Modern drug discovery does not slow down to wait for vocabulary to catch up. The next generation of therapeutics will demand a naming infrastructure that scales with the chemistry — one that grows its own vocabulary, audits its own decisions, and verifies every output against the molecule it describes. Connect with BIOVIA to explore how Helmsman, designed to evolve with the chemistry of modern therapeutics, can be your compass for monomer naming in this new age.
To learn more, read the recent article:

Dr. Arman Sadybekov is a Senior R&D Software Engineer at BIOVIA, based in San Diego, CA. For over a decade, Arman has been building cheminformatics and machine-learning methods for small-molecule drug discovery, and is currently responsible for predictive modelling in the Pipeline Pilot Chemistry SDK, fragment-based chemical-space search in BIOVIA Direct, and the computational chemistry capabilities behind Marie, BIOVIA's generative AI assistant on the 3DEXPERIENCE platform. His current focus is building agentic AI workflows for chemistry and training the AI chemists behind them. Arman has a Ph.D. in Theoretical Chemical Physics and an M.S. in Computer Science from the University of Southern California, USA, and an M.S. in Chemistry from Moscow State University, Russia.