
The chemical universe is vast. Modern make-on-demand combinatorial spaces have now reached the trillion-compound scale. Enamine’s xREAL Space, for example, includes over five trillion members. These libraries are great starting points for drug discovery projects, offering enormous chemical diversity and ready access to novel, drug-like molecules. But, how do you narrow those molecules down to a handful of candidates that are worth ordering and testing? BIOVIA Generative Therapeutics Design (GTD) answers this question by evolving molecules directly toward your objectives, while simultaneously constraining the search to compounds the library can actually deliver.
The Standard Approach: Screen the Building Blocks, Not the Library
Docking or QSAR-modeling every compound in a trillion-member chemical space against a target is practically impossible. Docking ten billion compounds at a typical rate would take over 3,000 CPU years, and evaluating a meaningful fraction of those, even with fast ML-based scoring remains expensive and time-consuming at this scale. In practice, leads must be identified in hours to permit cycles of testing and learning.
To address this challenge, many in the field have developed methods that exploit the modular nature of these libraries in a particular way. Rather than virtually testing a whole library of assembled products, some of the existing methods screen the individual building blocks first. These hierarchical approaches identify which scaffold (the core chemical framework) and synthon (a reactive building block fragment representing its contribution to the final product) combinations score best against the target in isolation, then enumerate, and dock only the subset of full molecules derived from those promising pairings. V-SYNTHES, for instance, can identify candidates while docking less than 0.1% of the total library.
While these methods are elegant, logical, and useful, there is still a significant computational expense associated with them, since the spaces include millions of synthons and trillions of compounds. Furthermore, once promising synthons are identified, many possible fully elaborated candidates allowed by chemistry of the specific chemical space have to be considered. Additionally, building-block-first methods are typically tied to a single scoring function, making it difficult to simultaneously optimize for aggregate properties such as selectivity and ADMET profile.
The BIOVIA Approach: Evolve Towards the Library
BIOVIA Generative Therapeutics Design (GTD) takes a fundamentally different approach. Rather than searching for matches inside the library that is approximated by building blocks, GTD evolves molecules toward a defined Target Product Profile (TPP) with guidance from a set of predictive models. The key innovation for combinatorial library targeting is a dedicated model that predicts whether a given molecule is likely to be a member of the specific library of interest. The process involves two independent, and thus modular and reusable, steps.
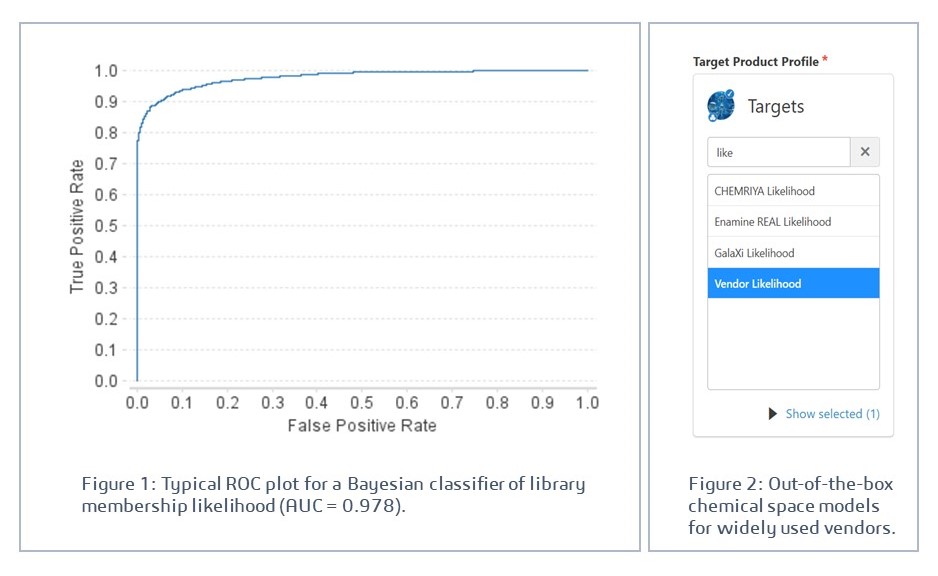
Step 1 — Build a library membership model
A machine learning model is trained to distinguish molecules that belong to the target combinatorial library from those that do not. Because such libraries are generated by a well-defined, finite set of chemical operations, on a relatively small collection of building blocks, this is a highly learnable problem, where the structural patterns that define library membership are consistent and tractable. The result is a reliable classifier that, given any candidate structure, returns a relative likelihood of library membership (Figure 1).
The 2026 version of GTD includes models for chemical spaces from leading vendors of make-on-demand molecules such as CHEMRIYA, Enamine, and WuXi. Additionally, a combined ‘Vendor’ model allows likely membership in any one of these collections as a satisfactory criterion when selecting a TPP (Figure 2).
Step 2 — Incorporate the model into a multi-objective TPP
The library membership model is added alongside the chemical structure constraints, target, anti-target and ADMET objectives when selecting a desired TPP. A desirability function is configured for its score, so that the candidates predicted to fall outside the library are penalized during the evolutionary search, steering the generator away from structures outside the selected library (or libraries!) without the need to enumerate a specific subset.
Why This Matters
The building-block-first strategy searches inside the existing library space and asks “which of these molecules scores best?” while the GTD strategy generates molecules guided toward the library and asks “what molecule best satisfies my objectives, and is it one I can actually buy?”. GTD evaluates whole molecules against whole objectives, no additivity assumption, and achieves multi-parameter optimization in a single run. Since the library membership model requires only a representative sample of library members to train, the same workflow also applies to in-house or proprietary combinatorial libraries, giving drug discovery teams a powerful way to mine their own chemical assets with the same efficiency.
GTD rapidly prioritizes a handful of candidates that can be ordered directly from a vendor with little computational expense. Rather than exhaustively searching for needles in a trillion-molecule haystack, GTD effectively steers you towards what you need, considering all your constraints and requirements. This reduces the time and cost associated with searching, and takes you straight to generating molecules to test, thus increasing the likelihood of success in your drug discovery projects.

Leo Bleicher is the Director of Bioscience and Scientific Informatics Development at BIOVIA in San Diego. He spent two decades working as a medicinal chemist, cheminformatics and drug discovery information scientist before joining BIOVIA in 2012.