


has reported 1,337,166 cases with 74,176 deaths throughout the world.
Examining SARS-CoV-2 at the genome level will provide insights into understanding the origins of this virus. It will also help scientists design diagnostic tools to detect this invisible pathogen and facilitate invention of therapeutics to minimize loss of life.
Understanding the SARS-CoV-2 Genome
A virus is an infectious agent that requires a living host to thrive and replicate. Also, SARS-COV-2 is a single-stranded RNA virus with a genome of nearly 30 kb nucleotide bases with 12 putative open reading frames. Shortly after the epidemic began in December of 2019, Chinese scientists sequenced the SARS-CoV-2 genome. Various scientific groups have released complete genomic sequences of SARS-CoV-2 in the last few weeks. These are publicly available in Genbank and the Coronavirus Database.
Origin of the SARS-CoV-2 Virus
During outbreaks such as this, non-scientific conspiracy theories can result in needless biases against countries, communities and cultures. SARS-CoV-2 is no exception, and the situation is only exacerbated by today’s mushrooming social media platforms. It is incumbent upon us to view this invisible enemy through a rational scientific lens. Based on genome analyses, SARS-CoV-2 is a virus that evolved naturally and is not a synthetic lab strain1,2. Scientists have sequenced the full genomes of more than 100 strains of SARS-CoV-2 collected from different regions of the world. It turns out that these strains are more than 99.5% identical on a nucleotide level. This indicates that the strains did not mutate much across different regions, ostensibly as the virus already has a high infection rate and virulence.
In the recent past, two other coronaviruses have received global attention. These were the SARS-CoV, China, 2002, and the MERS-CoV, Saudi Arabia, 2012. Both of these earlier viruses were shown to have originated in bats. Based on this historical knowledge, scientists sequenced the coronavirus from the bats and showed that Bat CoV (RaTG13) was 96.2% identical to SARS-COV-2, thus confirming the zoonotic origin of the latter.2 The coronavirus often uses an intermediate carrier before infesting humans. Interestingly, around Oct 2019 reports of dead Malayan Pangolins with lungs and pulmonary frothy fibrosis symptoms at Guangdong Wildlife Rescue center in China prompted scientists to isolate their metagenome. Indeed, the metagenome data from the dead pangolins contained the coronavirus! 3
Interestingly, at the whole genome level, SARS-CoV-2 is nearly 91% identical to Malayan Pangolin CoV, indicating that Pangolins could be an intermediate host.
What are Pangolins? They are ant-eating mammals that are in high demand in Asia for use in traditional Chinese medicine as well as for their meat, which many consider a delicacy. They are also today’s most trafficked mammal in the illegal wildlife trade.
SARS-CoV-2 is different from other known coronaviruses, with 88% or less sequence identity. Based on phylogenetic analyses, SARS-CoV-2 seen in humans, bats (RaTG13) and Malayan Pangolins is a novel class of beta coronavirus. Nearly 35 different types of coronavirus strains from different parts of the world and from different organisms have been analyzed at the whole genome level. SARS-CoV-2, shown in blue below, is a novel class of beta coronavirus (Figure1).
As previously noted, the Spike protein contains two functional domains: a receptor binding domain and a second domain which contains sequences that mediate fusion of the viral and cell membranes. The Spike glycoprotein must be cleaved by cell proteases to enable exposure of the fusion sequences and hence is needed for cell entry. Comparison of the S1/S2 cleavage site sequence from Pangolin CoV and bat-SARS-CoV-2 shows an insertion of the furin recognition motif. This indicates a distinct mechanism for entry of the viral genome into the host cytoplasm for replication as shown in Figure 3.
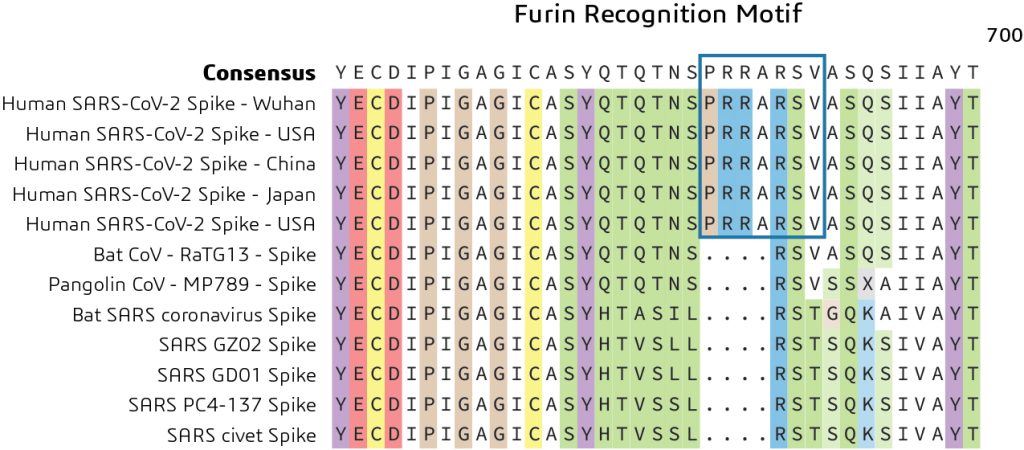
What is the role of the furin recognition motif? In humans, the furin recognition motif (PRRARSV) is recognized by the FURIN protein, a member of the S8 family of subtilisin-like peptidases that helps to remove sections of the protein to change their conformation from an inactive to an active state.
It has been suggested that the acquisition of this furin cleavage site might be a ‘gain of function’ that enabled a bat CoV to jump into humans and begin its current epidemic spread. This might be a potential avenue for exploring novel drugs targeting the blocking of this motif to prevent the replication of the virus inside the host.
Thus, careful examination of the Spike protein in SARS-CoV-2 shows the optimized RBD, a furin recognition motif, like some MERS coronaviruses, and its ability to bind to the ACE2 protein strongly. This suggests a natural selection process in play. Natural recombination events in viruses co-infecting a host have been shown to improve their host range, while also increasing virulence and virus adaptation. SARS-CoV-2 genome data with backbone of the bat (RaTG13) and pangolin CoV again indicate that this is a virus generated by natural recombination.
What Is the Immediate Donor of SARS-CoV-2 to Humans?
The SARS-CoV-2 sequence has a mix of both bat-SARS-CoV (RaTG13) as well as regions of conserved Pangolin CoV that can only happen during recombination of these viral genomes. Also, a gain of function, as seen with the furin recognition motif involves another virus recombination. For recombination to occur, it is only logical that there should be a natural host that harbors these viral genomes. Is it another pangolin? Or another wild animal in the Wuhan sea food market? This is still unknown. Understanding the origin could help to prevent future outbreaks of viral strains and global pandemics.
For more information, please contact us.
Reference
- The proximal origin of SARS-CoV-2. Andersen, KG, Rambaut A, Lipkin, WI, Holmes, EC and Garry, RF. Nature Medicine(2020), 17th March, 2020
- https://www.livescience.com/coronavirus-not-human-made-in-lab.html
- Probable Pangolin Origin of SARS-CoV-2 Associated with the COVID-19 Outbreak. Zhang T, Wu Q, Zhang Z. Curr Biol. 2020 Mar 13. pii: S0960-9822(20)30360-2. doi: 10.1016/j.cub.2020.03.022
- Genomic variance of the 2019-nCoV coronavirus. Ceraolo C, Giorgi FM. . J Med Virol.2020 May;92(5):522-528. First published:06 February 2020. https://doi.org/10.1002/jmv.25700