
Materials Studio offers a user-friendly yet powerful platform for modeling a huge range of systems, with this report focusing specifically on polymers and polymer networks. In conjunction with Pipeline Pilot, various methods are available to model such complex materials ranging from the widely reported cross-linked polyethylene (XLPE), to custom networks with unique reaction mechanisms. United atom forcefields are developed to efficiently model such systems involving large numbers of particles, providing a balance between computational efficiency and accuracy.
The Importance of Forcefields in Molecular Simulation
A critical element of molecular simulation is the forcefield. Forcefields govern the behaviour of particles in a system through both bonded and non-bonded interactions, covering contributions including bond stretching, angle bending, dihedral torsions, etc. In polymer models, the forcefield is essential for determining how polymer chains fold, entangle with one another and respond to external stimuli. By accurately modelling these atomic-level interactions, forcefields enable the prediction of macroscopic properties such as density, radius of gyration, and glass transition temperature. Thus, an effective forcefield is essential for both the accuracy of simulations and the reliability of the predicted properties.
There are a number of forcefields available depending on the type of system being modelled. Some forcefields are optimized for specific materials while others, like COMPASSIII [1], are designed for a broader range of materials. The field of forcefield development is continually developing as exemplified by the recent emergence of MACE [2]; a machine learning software, which is used, among other things, to generate forcefields. Of particular significance in polymer modelling are OPLS and OPLS-UA. OPLS, Optimised Potentials for Liquid Simulations [3], was specifically designed to simulate liquids and later expanded to cover a wide range of organic molecules, biomolecules, and polymers. OPLS-UA is a modified version of this forcefield for a united atom approach. Given that molecular simulations can be computationally expensive, especially for systems with large numbers of particles, it is often desirable to simplify these systems by reducing particle counts while simultaneously minimizing the loss of accuracy. This simplification is achieved through united atom or coarse-graining methods These approaches also provide a further benefit of longer time steps, allowing users to conduct simulations with more steps within a set timeframe.
Custom Forcefield Development in Materials Studio
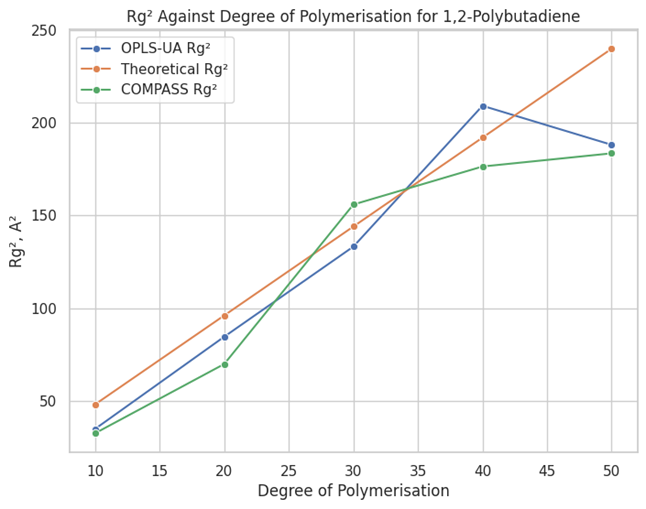
In Materials Studio, users create custom forcefields by inputting parameters in various functional forms, gaining full control. Through literature review and modification of OPLS atomistic parameters, followed by iterative testing and refinement, an OPLS-UA custom forcefield was developed [4]. Torsion parameters, for instance, were validated using conformational analysis on single molecules. The integration of Perl scripting within Materials Studio provided precise control, and a simple script was created to extract torsion energy as a function of torsion angle. These results were then compared to literature data or DMol³calculations. Once refined, the forcefield was tested on six polymer systems, with an Amorphous Cell constructed for each system and equilibrated through molecular dynamics simulations. Physical properties of the equilibrated systems were extracted and compared to literature values. One key test was the radius of gyration as a function of the degree of polymerization, which is well documented for many polymers. To automate the process of generating cells, equilibration, and calculating the radius of gyration for systems with incrementally larger degrees of polymerization, custom Pipeline Pilot protocols were developed [5]. This integration significantly reduced time and minimized human error. It allowed for the radius of gyration data to be compared with theoretical values and ultimately validate the accuracy of the developed forcefield, as exemplified in Figure 1.
Alongside the creation and validation of the forcefield, a simple method was developed to convert a polymer from full atomistic to a united atom representation. This was achieved by removing hydrogen atoms, adjusting the mass numbers of the corresponding beads, and assigning the appropriate forcefield type to each bead. A custom Perl script was created for this process, using a Study Table to perform a pattern search through the included fragments. Each fragment corresponds to a different atom type, such as sp³ CH₂, sp² CH, or aromatic CH, along with their associated forcefield types. As a result, only the name of the input 3D Atomistic Structure file needs to be inserted into the script for the polymer to be automatically converted to united atom and typed.
With a method in place to convert and type polymers, along with the validated OPLS-UA forcefield, polymer and polymer network modelling using this forcefield could be further explored. Depending on the system being constructed, various reactions need to be modelled—most notably addition, cycloaddition, and condensation reactions. Materials Studio offers several methods for this purpose, with the choice depending on the central reaction. Three main methods were employed to model these systems: a customizable networking Perl script, a Pipeline Pilot protocol, and the Reaction Finder tool. Examples of each are discussed below.
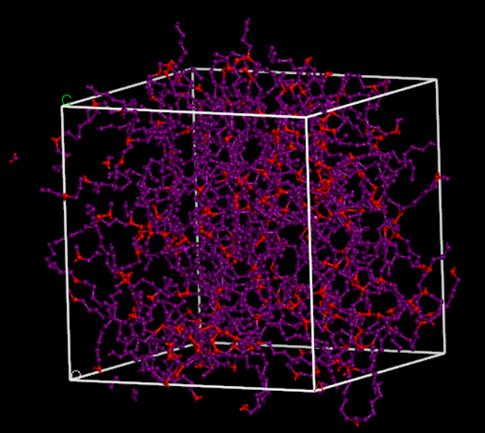
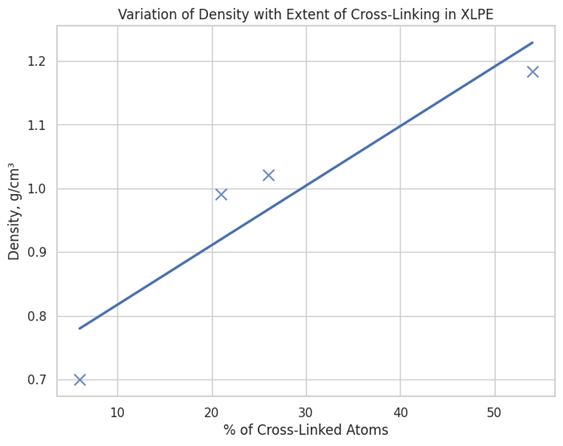
Cross-linked polyethylene (XLPE) is a widely studied polymer network, used in numerous applications ranging from electrical insulation in wires to components in fuel gaskets, owing to its high chemical and heat resistance. Users can model this network in Materials Studio using the Pipeline Pilot connector with the modified Network Protocol, as demonstrated in Figure 2. The process begins by creating an Amorphous Cell containing both ethane molecules and polyethylene chains, with reactive atoms defined. These reactive atoms dictate the reactions that build the network, allowing the polymer chains to grow linearly, branch out, or cross-link with other chains. The probability of each reaction is specified, providing additional control over the end system. By altering the probability of the defined reactions, cross-linking density is modified and the effect on physical properties after equilibration can be investigated, as exemplified in Figure 3.
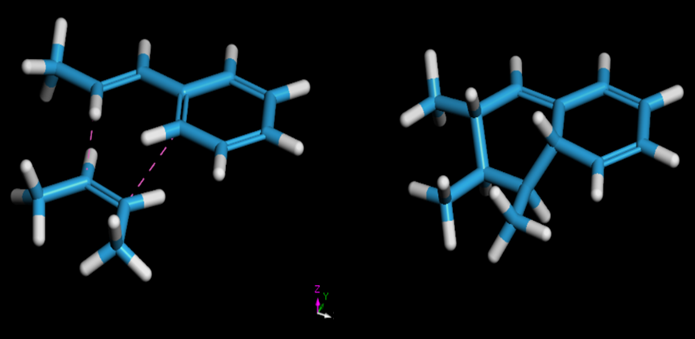
Diels-Alder reactions are prominent cycloaddition reactions between a diene and an alkene, and can be used to join monomers in more complex polymer systems. The Reaction Finder[6] tool is employed to precisely define these reactions by drawing both the reactants and products, mapping the atoms between them, and identifying close contacts between the reactive atoms. This is exemplified in Figure 4. This approach offers significant control, making it particularly useful for modelling specific structures with atypical reactive sites.
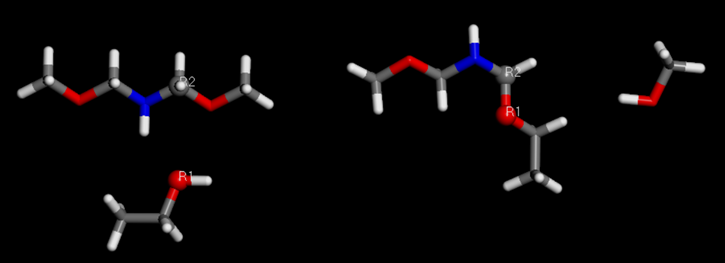
To model condensation reactions, a customizable networking Perl script[7] was adapted and used effectively. Reactive atoms are defined in both the monomer and curing agent, which are then organised into an Amorphous Cell. The script specifies these reactive atoms along with the input structure document. An additional subroutine within the script is used to define specific bond-breaking events during the reaction, producing the desired product as shown in Figure 5. This versatile script supports the cross-linking of various monomers and curing agents and can be tailored to meet different system requirements.
Conclusion: The Power of Materials Studio for Modeling Polymer Networks using United Atom Forcefields
In conclusion, Materials Studio proves to be an exceptional platform for modelling complex polymer systems and networks, offering a wide range of tools and customization options. Its integration with Pipeline Pilot, the ability to develop and refine custom forcefields, and the flexibility provided through Perl scripting enable precise control over model building and simulations. The platform’s versatility and the high degree of control it offers open up exciting possibilities for future research, allowing users to push the boundaries of polymer design, optimization, and property prediction with great accuracy and efficiency.
[1] Akkermans, R. L. C., Spenley, N. A. and Robertson, S. H. (2020) ‘COMPASS III: automated fitting workflows and extension to ionic liquids’, Molecular Simulation, 47(7), pp. 540–551, doi: 10.1080/08927022.2020.1808215.
[2] https://mace-docs.readthedocs.io/en/latest/ (accessed 09/2024).
[3] William L. Jorgensen, David S. Maxwell, and Julian Tirado-Rives (1996), ‘Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids’, Journal of the American Chemical Society , 118 (45), pp. 11225-11236, doi: 10.1021/ja9621760.
[4] Available in the BIOVIA Materials Studio community.
[5] Available in the BIOVIA Materials Studio community.
[6] J.W. Abbott and F. Hanke, ‘Kinetically Corrected Monte Carlo-Molecular Dynamics Simulations of Solid Electrolyte Interphase Growth’, J Chem Theor Comput, 18, 925 (2022).
[7] Developed by Jason DeJoannis, Stephen Todd & James Wescott (available in the BIOVIA Materials Studio community).

Sophie Whitaker completed her Master’s degree in Chemistry at the University of Oxford, specializing in energy materials chemistry. She is currently an intern with the BIOVIA team at Dassault Systèmes, where her work focuses on united atom forcefield development and utilizing Materials Studio and Pipeline Pilot to model polymer networks.