
Understanding Molecular Crystals and Their Computational Study
Molecular crystals, such as trinitrotoluene, paracetamol or caffeine1, are composed of individual molecules bound by nonbonded interactions2. Intramolecular bonds that hold the atoms within each molecule are much stronger than the intermolecular bonds that bind the molecules together in a crystal. The cohesive forces that bind these molecules may include Van der Waals forces, dipole-dipole interactions, π-π interactions and hydrogen bonding. Furthermore, their properties are closely related to their structure making accurate prediction of crystal structures and properties a crucial challenge in various industries. Researchers study inorganic and organic crystals to develop innovative drugs and energetic materials.
Moreover, computational chemistry has emerged as a valuable tool, enabling the rapid study of these compounds3. By using mathematical models and equations various molecular properties and characteristics can be predicted prior to their synthesis in a lab, minimizing the cost, time and risks associated with their synthesis. Crystal prediction4, including lattice energy prediction, density calculations, and polymorphism prediction are extensively studied, particularly in the pharmaceutical and energy industries.
Forcefields5,6 (or interatomic potentials) are particularly interesting computational models used in classical simulations for studying, among others, crystal structures. Over the years, researchers have developed several forcefields to accurately predict their properties. Moreover, comparative studies have been conducted to evaluate these forcefields through different protocols. Robinson et al.6 presents an extensive comparison of 324 forcefield protocols for calculating lattice energies of molecular crystals. This study examined several well-known forcefields, including DREIDING, Universal, CVFF, PCFF, and COMPASS, using the Materials Studio® collection They highlighted the COMPASS (Condensed-phase Optimized Molecular Potentials for Atomistic Simulation Studies) forcefield for its effective performance in lattice energy calculations on general organic molecular crystals, after analyzing 235 crystals from the Cambridge Structural Database7. The study demonstrated that several protocols based on this forcefield achieved good performances and relatively low standard deviation.
COMPASSIII for Molecular Crystals
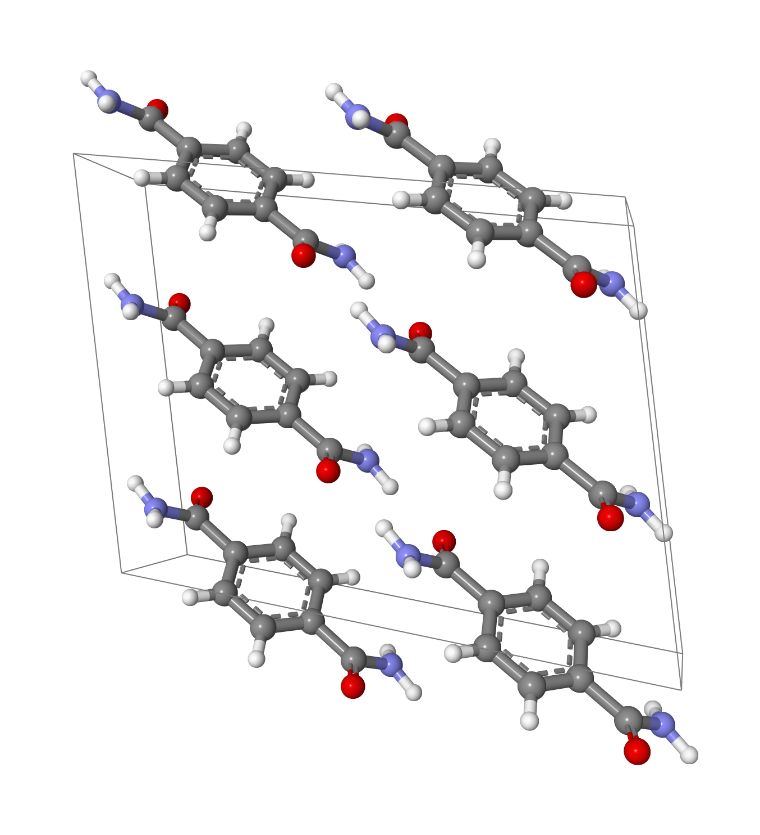
To further improve COMPASS, the Materials Studio Cambridge team developed COMPASSIII8, an extended version for ionic liquids and other materials of the COMPASS9 forcefield, originally designed for condensed-phase properties predictions with a focus on materials. It was the first ab-initio forcefield that enabled accurate and simultaneous prediction properties for a broad range of molecules. This forcefield largely inherited its structure from the Consistent Force Field (CFF). The new version COMPASSIII introduced 27 new types, covering a wide range of materials, with their own typing rules and valence interactions.
To assess the accuracy of COMPASSIII to predict lattice energies and densities of molecular crystals, calculations were performed, using the Forcite module of Materials Studio 2024, on the 235 structures used by Robinson et al6. To automate calculations and make the analysis more efficient, Pipeline Pilot®10 protocols, and in particular the Materials Collection, were utilized, allowing to consecutively run simulations on all desired molecules. Spearman’s correlation coefficient, R-squared value and Root Mean Squared Error (RMSE) were calculated using experimental data from the Cambridge Structural Database. The results, presented in Table 1, demonstrate that COMPASSIII yields enhanced results compared to various forcefields available in Materials Studio.
Table 1: Result comparison for lattice energy calculations on 235 crystals using, COMPASSIII, Universal, DREIDING, CVFF and PCFF forcefields
| COMPASSIII | Universal | DREIDING | CVFF | PCFF | |
| R2 | 0.70 | 0.29 | 0.40 | 0.32 | 0.51 |
Lattice energy and density calculations with COMPASSIII were also performed on a larger dataset containing 821 molecules, which included the structures used by Robinson et al6. Density, another significant property of molecular crystals, was accurately predicted with COMPASSIII, achieving an R-squared of 0.97, as shown in Table 3. Although the lattice energy results were promising, as shown in the previous table, there is still potential for further enhancement. To enhance these results, outliers, corresponding to compounds with relative error for lattice energy over 10%, were identified and further investigated. The main problematic family of compounds was those with amino and nitro functions on aromatic heterocycles.
Table 3: Results for lattice energy and density calculations on 821 molecular crystals using COMPASSIII forcefield
| Lattice Energy | Density | |
| Spearman | 0.87 | 0.99 |
| R2 | 0.74 | 0.97 |
| RMSE | 17.6 kJ/mol | 0.04 g/cm³ |
Study of Compounds to Enhance COMPASSIII Parameters
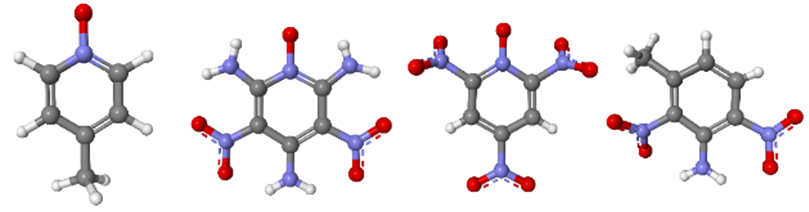
To improve the COMPASSIII forcefield, problematic compounds were examined, particularly those with amino and nitro functions on aromatic heterocycles. Among these, there are some complex structures, such as 2,6-Diamino-3,5-dinitropyrazine-1-oxide, also known as LLM-105. This molecule, featuring a pyrazine-N-oxide structure with two amino and two nitro substituents, is highly energetic. This material is fascinating because of its great thermal stability and insensitivity to friction, impact and shock. This molecule, as well as simpler ones containing similar chemical functional groups, were studied. In particular, partial charges were recalculated with Density Functional Theory (DFT) using the Electrostatic Potentials (ESP) method in DMol3, and incorporated in COMPASSIII.
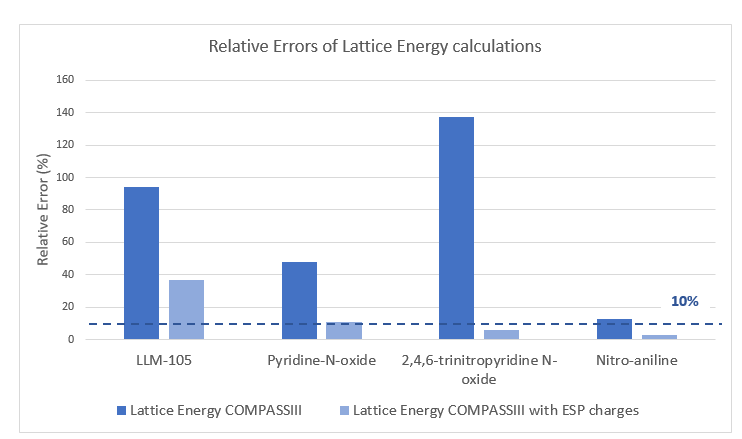
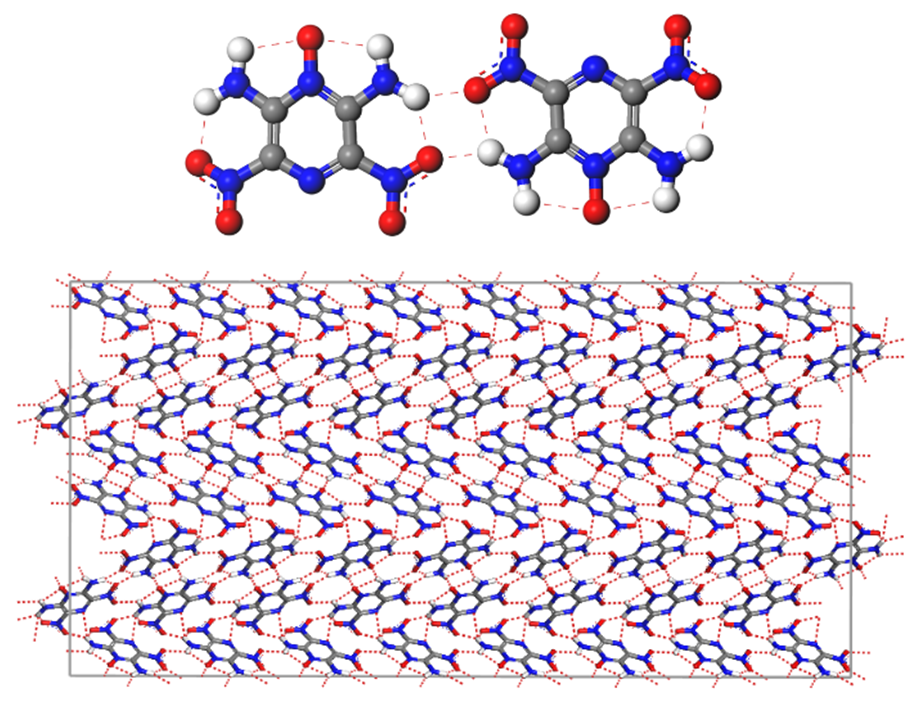
For most structures with a high relative error in lattice energy, adjusting the partial charges used by COMPASSIII reduced the error below the 10% threshold, as shown in Figure 3. However, while adjusting the charges significantly decreases the error, LLM-105 presents more complexity and needs further improvement. Indeed, the molecular crystal of LLM-105 has a significant hydrogen bonding network, displayed in Figure 4, which can influence lattice energy and density computations necessitating further investigations.
Conclusion
Accurately predicting the properties of molecular crystals is a significant challenge. As previously demonstrated, the COMPASSIII forcefield outperforms other well-known forcefields for these compounds. However, it is essential to continually optimize and review forcefields to ensure accurate parametrization and simulations. By continuously improving COMPASSIII, precise predictions for a wide range of compounds can be achieved.
Learn more about BIOVIA Software.
References
(1) Braga, D., Casali, L., & Grepioni, F. (2022). The relevance of crystal forms in the pharmaceutical field: sword of damocles or innovation tools?. International Journal of Molecular Sciences, 23(16), 9013.
(2) Politzer, P., & Murray, J. S. (2003). Energetic materials: part 1. Decomposition, crystal and molecular properties. Theoretical and Computational Chemistry, Volume 12. Elsevier: Amsterdam.
(3) Jorgensen, W. L., & Tirado–Rives, J. (2005). Molecular modeling of organic and biomolecular systems using BOSS and MCPRO. Journal of Computational Chemistry, 26(16), 1689-1700.
(4) Nyman, J., Pundyke, O. S., & Day, G. M. (2016). Accurate force fields and methods for modelling organic molecular crystals at finite temperatures. Physical Chemistry Chemical Physics, 18(23), 15828-15837.
(5) Ermer, O. (2005). Calculation of molecular properties using force fields. Applications in organic chemistry. In Bonding forces (pp. 161-211). Berlin, Heidelberg: Springer Berlin Heidelberg.
(6) Marchese Robinson, R. L., Geatches, D., Morris, C., Mackenzie, R., Maloney, A. G., Roberts, K. J., … & Vatvani, D. R. M. (2019). Evaluation of force-field calculations of lattice energies on a large public dataset, assessment of pharmaceutical relevance, and comparison to density functional theory. Journal of chemical information and modeling, 59(11), 4778-4792.
(7) Groom, C. R., Bruno, I. J., Lightfoot, M. P., & Ward, S. C. (2016). The Cambridge structural database. Structural Science, 72(2), 171-179.
(8) Akkermans, R. L., Spenley, N. A., & Robertson, S. H. (2021). COMPASS III: Automated fitting workflows and extension to ionic liquids. Molecular Simulation, 47(7), 540-551.
(9) Sun, H. (1998). COMPASS: an ab initio force-field optimized for condensed-phase applications overview with details on alkane and benzene compounds. The Journal of Physical Chemistry B, 102(38), 7338-7364.
(10) BIOVIA, Dassault Systèmes, Pipeline Pilot, version 2024, San Diego: Dassault Systèmes, 2024.

Amély Rondeau is a Chemical Engineering student at the European School of Chemistry Polymers and Materials in Strasbourg specialized in materials science and artificial intelligence. She is an intern at Dassault Systèmes, where she applies her research skills as in the BIOVIA team. During her internship she works on forcefield optimization for crystalline system studies and the use of Machine Learning for forcefield implementation.