Understanding how proteins communicate is like deciphering a complex molecular telephone game. When a ligand binds to one site on a protein, it can trigger conformational changes that propagate through the structure, affecting function at entirely different locations. This intricate phenomenon—known as allosteric communication—holds the key to designing more effective therapeutics and understanding protein function at a deeper level.
Think of breathing – as you breathe, a series of allosteric interactions take place in your body orchestrated by the well-known oxygen-carrying protein hemoglobin. When oxygen attaches to one subunit of hemoglobin in your lungs, it causes conformational changes in the other subunits, allowing them to grab oxygen more easily. When hemoglobin reaches tissues that need oxygen, releasing oxygen from one subunit makes it easier for the other subunits to let go as well. This allostery makes hemoglobin an effective mechanism to transport the oxygen, perfectly illustrating how binding at one site can influence what happens at completely different locations within the same protein.
While hemoglobin’s allosteric behavior has been well-studied and understood, detecting and mapping such communication networks in other proteins remains one of the most challenging puzzles in modern drug discovery and protein engineering. The problem is that these communication networks are incredibly subtle and complex, with most traditional methods lacking the sensitivity to detect these hidden connections between distant protein sites. Before the arrival of advanced molecular dynamics (MD) simulations combined with trajectory analysis tools, scientists relied heavily on static structural comparisons and limited experimental techniques that often missed the critical dynamic components of allosteric communication.
Over the last decade, this has been changing with the development of comprehensive computational workflows that can systematically trace how binding events ripple through molecular structures, revealing hidden communication networks that were previously invisible to researchers.
🎬 Watch the video to learn more:
Revolutionizing Allosteric Detection with Advanced Analysis Tools
At its core, modern allosteric analysis leverages sophisticated computational approaches that go far beyond simple structural comparisons. Starting from dynamic trajectories that capture proteins in motion over time, advanced analysis tools then meticulously extract meaningful patterns from these complex datasets through a series of iterative steps. In each step, they refine our understanding of how different protein regions communicate, guiding researchers towards coherent, biologically relevant insights into allosteric networks.
This powerful analytical capability opens doors to tackling a wide array of therapeutic design challenges. For example, simulation trajectory analysis can be employed to identify novel allosteric sites engineered for drug targeting. In the realm of protein-protein interactions, it can reveal new regulatory mechanisms aimed at optimizing binding specificity or enhancing therapeutic selectivity.
Comprehensive Multi-Step Workflow for Allosteric Analysis
The journey from raw trajectory data to meaningful allosteric insights involves several key computational steps that work synergistically to provide a complete picture of protein dynamics and druggability.
Feature data can be extracted from trajectories through two complementary approaches. The Measure Trajectory Features protocol extracts traditional structural parameters from complex simulations, capturing essential dynamic properties including inter-residue distances and backbone flexibility. Alternatively, the Detect and Analyze Pockets protocol can serve as the primary feature extraction tool, focusing specifically on cavity-related properties throughout the trajectory.
The Detect and Analyze Pockets protocol represents a crucial component of the allosteric analysis pipeline, utilizing mdpocket, a well-regarded pocket detection algorithm specifically designed for molecular dynamics trajectories, to identify and characterize binding sites throughout protein simulations. Unlike static pocket detection methods that analyze single conformations, mdpocket tracks pocket evolution over time, revealing how cavities open, close, and reshape during protein dynamics. This temporal perspective is essential because regulatory sites often exist as transient or cryptic pockets that are invisible in static structures but become accessible during conformational transitions.
The protocol provides comprehensive dynamic pocket tracking by monitoring their properties including volume, surface area, depth, and hydrophobicity throughout the entire trajectory, giving detailed insight into how binding sites respond to conformational changes and ligand binding events. Perhaps most importantly for therapeutic applications, the protocol incorporates advanced druggability assessment algorithms that evaluate each detected pocket’s potential as a drug target, considering factors such as persistence, size distribution, hydrophobic content, and geometric complementarity to known drug-like molecules. This opens new avenues for drug design, enabling researchers to understand how pocket dynamics correlate with broader allosteric networks throughout the protein structure.
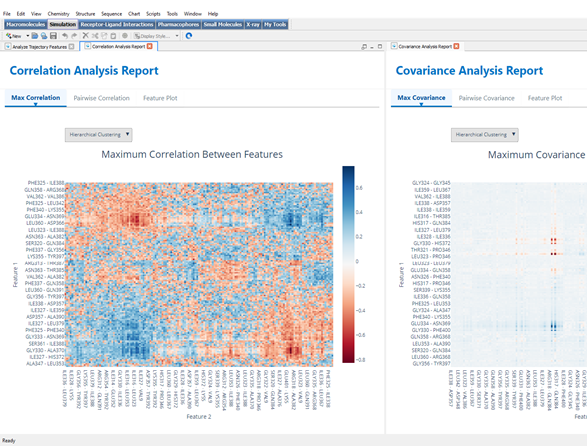
The extracted feature data from either protocol is then processed by the Analyze Trajectory Features protocol, powered by cross-correlation/covariance calculations that reveal dynamic coupling patterns between distant protein regions. This analysis can incorporate data from traditional residue-level features, pocket dynamics, or a combination of both approaches, providing a comprehensive view of long-range communication.
The analysis incorporates effective visualization techniques like hierarchical clustering, which computationally groups features with similar dynamic behaviors. This validation step ensures that identified networks represent genuine allosteric communication pathways rather than statistical noise, with particular attention to correlations between pocket dynamics and conformational changes.
Integrated Workflow Implementation
This comprehensive workflow—from initial feature extraction to final network validation—is available in BIOVIA Discovery Studio Simulation in the 3DEXPERIENCE® Cloud in 2025. The integration of pocket detection and characterization with traditional trajectory analysis transforms the complex challenge of allosteric detection into a significantly more manageable and effective task for researchers.
The combined approach enables researchers to not only identify allosteric communication pathways but also assess their therapeutic potential through integrated druggability analysis. This advances structure-based drug design, providing a direct path from fundamental allosteric discovery to practical therapeutic development strategies.
📩Want to find out the latest news about BIOVIA events, customer stories, blogs and more?
 Defne Ozgulbas
Defne OzgulbasDr. Defne G. Ozgulbas is an R&D Software Engineering Specialist in Protein Modeling and Simulation team at BIOVIA, Dassault Systèmes. She received her Ph.D. in Biophysics and Computational Biology from the University of Illinois at Urbana-Champaign at the Theoretical and Computational Biophysics Group, where she was awarded the Beckman Graduate Fellowship. Her doctoral research focused on biomolecular modeling, protein design and engineering, and protein-membrane interactions using advanced molecular dynamics simulations. At BIOVIA, Defne develops modeling and simulation tools in drug discovery and development.
 Tien Luu
Tien LuuTien Luu is the Product Manager and Principal Scientific Specialist at BIOVIA Dassault Systèmes, managing aspects of life science modeling and simulation predictive science solutions. She began her career as a Scientific Specialist after receiving her Ph.D. in Chemistry from the University of Wollongong, Australia. Since joining BIOVIA 22 years ago, Tien’s passion and excitement for helping customers accelerate their R&D have been steadfast and she is committed to improve customer experiences and product successes. In addition, she continues to be an active leader in scientific software product development, new program initiatives, and new applied and fundamental scientific research.