

After pharmaceutical scientists identify a lead compound, but before clinical trials, they must decide on the final formulation of the active pharmaceutical ingredient (API). This requires identifying the best solid form for the API and its potential excipients, so that the formulated product is ready for use by patients. This pre-formulation stage of development lasts about 2 years and aims to assess all solid-state properties of the API, such as crystal structure, solubility, morphology, etc. The pre-formulation stage also allows scientists to anticipate and identify any potential manufacturing issues, which could be costly to fix if identified later in the process (see e.g., Abbott’s Ritonavir case (1)).
Large pharmaceutical companies are becoming increasingly aware that an ‘in silico first’ approach is hugely valuable across the development phases. The computational investigation of potential drugs makes it possible for scientists to identify physical properties prior to experimental testing which can accelerate drug development, reduce costs and conserve laboratory resources. This move to virtual testing is resulting in a lower number of manufacturing issues and overall shorter time-to-market.
Optimizing Pharmaceutical Formulations
BIOVIA Dassault Systèmes offers many research tools that pharmaceutical scientists can use to perform such in silico risk assessment and experimental preparation. With BIOVIA’s state-of-the-art visualization tools, you can easily import and manipulate crystallographic data prior to computing physical properties such as stability and solubility. Most notably, you can predict crystal structures based on the chemical representation of the API and its powder diffraction pattern (XRPD) using advanced Rietveld-based techniques. Scientists are constantly assessing the water solubility of drugs, because low water solubility can result in poor bioavailability. Various in silico methods offered by BIOVIA are available to formulators for this use, ranging from in-house developed QSPRs to tools based on COSMO-RS that are more computationally demanding. You can also use simulation tools to address properties that impact shelf lifetime, like chemical or physical drug stability. For example, you can predict glass transition temperature or degradation pathways or mechanisms.
BIOVIA Science Council’s current research focusses on employing a series of molecular modelling methods to predict the effect of solvents on (crystal habits) Crystal Growth and Morphology. Today we use many different levels of theory to study pharmaceutical ingredients and formulations, from classical mechanics solving Newton’s equations of motion to the application of quantum mechanical tools that aim to solve Schrödinger’s equations for the system studied and COSMO-RS solution thermodynamics.
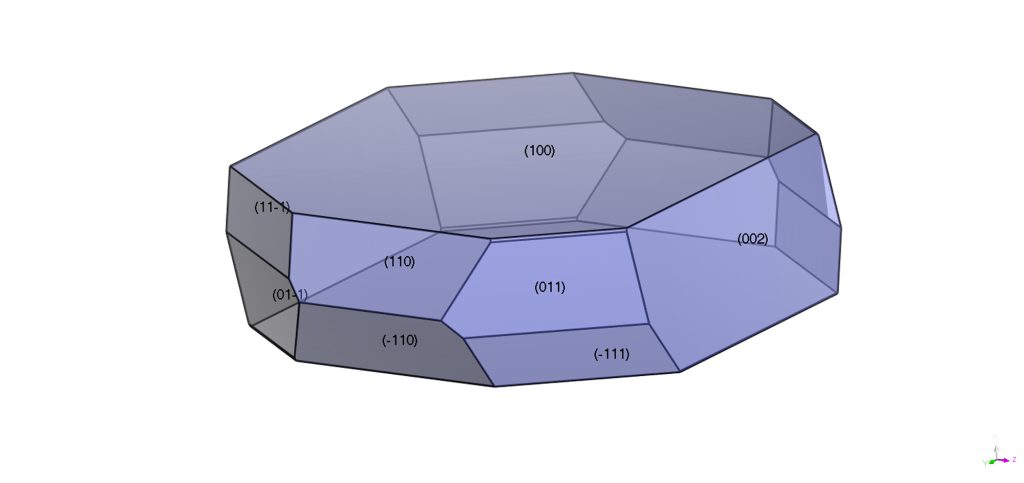
Leveraging Crystal Design and Engineering
Computer simulations are increasingly employed to predict the morphology of crystals. (2) Controlling the shape of crystals is one of the primary goals of crystal design and engineering. Many physical properties that are relevant to pharmaceutical development such as solubility, tabletting, compressibility and dissolution rate depend on the morphology of the crystal. (3) Needle-like crystals are undesirable as they are brittle and do not possess the shear strength to withstand the tabletting process. Understanding the chemical properties of the crystal faces provides opportunities to control the growth process and produce crystals with more isometric morphology.
Of course, the internal chemical structure is not the sole determinant of Crystal Growth and Morphology, which also depends on external factors such as solvents (pure or a mixture of), supersaturation and impurities. (4) The effect of solvents can be significant, particularly if the solvent interacts differently with each surface of the crystal. The choice of solvent for crystallization of a given substance is therefore very important.
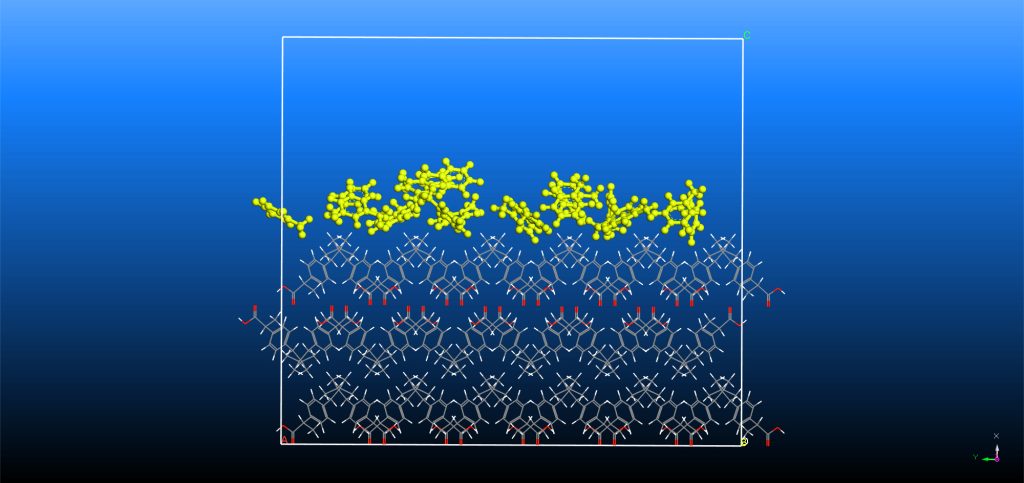
Computational Simulation Methods
We can employ several computational simulation methods when analyzing an API’s crystal system. Crystal Growth and Morphology
The Materials Studio Morphology tool enables prediction in vacuum of the minimum energy crystal structure and identification of the key crystal surfaces of an API. Monte-Carlo algorithms with BIOVIA’s COMPASS III force-field (5) can subsequently estimate the adsorption binding energy of a single molecule of solvent, impurity or additive onto a crystal surface and, in turn, we can predict its potential effect on crystal growth. (6)
Analyzing the dynamic evolution of a crystal model using molecular dynamics provides insight into the molecular interactions of a system. We can perform simulations of a solvent layer on the morphologically important faces to estimate the crystal-solvent interaction energy. Although computationally more expensive, the modified attachment energy (MAE) algorithm can improve the prediction of morphology and aid identification of undesirable needle-like crystals. (7)
In Silico versus Trial & Error
Adopting an in silico approach in the early drug development stages is more efficient than the traditional culture of a trial-and-error approach, while also offering extensive development opportunities for the pharmaceutical sector. The industry can use modelling techniques such as those mentioned in this article for fast initial drug formulation screening. Computational simulations are much faster than their experimental equivalent and save limited and valuable drug resources.
Our goal is to develop a tool that facilitates the selection of suitable crystallization solvents, while also providing crystallization scientists with a starting point for further process optimization of supersaturation, temperature and impurities levels. A fundamental understanding of materials properties leads to faster, more effective pharmaceutical development. To learn more about in silico approaches to drug development, see the on-demand BIOVIA Science Council webinar on Augmenting Pharmaceutical Development with In Silico Materials Design.
Learn More about Materials Studio
References
-
Bauer, S. Spanton, R. Henry, J. Quick, W. Dziki, W. Porter and J. Morris, Pharm. Res., 2001, vol. 18, 859–866
-
L. Rohl, Curr. Opin. Solid State Mater. Sci., 2003, 7, 21–26.
-
Bisker-Leib and M. F. Doherty, Cryst. Growth Des., 2001, 1, 455–461.
-
Chen, M. Xia, W. Lei, F. Wang and X. Gong, J. Mol. Model., 2013, 19, 5397–5406.
-
L. C. Akkermans, N. A. Spenley and S. H. Robertson, Mol. Simul., 2013, 39, 1153–1164.
-
T. Konkel and A. S. Myerson, Mol. Simul., 2008, 34, 1353–1357.
-
Chen and B. L. Trout, in Crystal Growth and Design, American Chemical Society, 2010, vol. 10, pp. 4379–4388.